Epidemiological studies show no clear relationship between hypermobility and widespread pain. This questions the validity of diagnoses such as hypermobile spectrum disorder (HSD) and hypermobile Ehlers-Danlos Syndrome (hEDS). Based on its prevalence, symptom presentation, female predominance, and lack of molecular basis, hEDS resembles fibromyalgia and ME/CFS more than other EDS types. The British Society of Paediatric and Adolescent Rheumatology expressed concern that ascribing symptoms to HSD/hEDS without evidence of connective tissue disease is unlikely to be helpful and may cause harm.

The odd one out
Ehlers-Danlos Syndrome (EDS) is a group of heritable connective tissue disorders characterized by joint hypermobility, skin hyperextensibility, and tissue fragility. The most recent diagnostic criteria, published in 2017, identify 13 EDS subtypes. Twelve have a known genetic mutation involving collagen synthesis or other extracellular matrix proteins. These are (very) rare conditions with an identified molecular basis and prominent clinical signs. Classical EDS, for example, is known for hyperextensible and fragile skin and has an estimated prevalence of 1 out of 20,000 people (0.005%). Vascular EDS increases the risk of arterial ruptures and occurs in only 1 in 90,000 people (0.001%). Other EDS types are even rarer with features such as an abnormal curvature of the spine, spontaneous lung collapse, or rupture of the cornea in the eye.
The odd one out is hypermobile EDS (hEDS). Out of 13 EDS types, it is the only one that does not have a known molecular basis or genetic mutation. With an estimated prevalence of 1 out of 5,000 (0.020%), it is much more common than other EDS types. The vast majority (approximately 80-90%) of EDS patients are believed to have hEDS.
Yet despite hEDS dominating the EDS population, it is also considered the most controversial diagnosis. It is characterized by non-specific features such as joint hypermobility and widespread pain that are common in the general population. Rather than by specific clinical signs, hEDS stands out by its wide range of debilitating but poorly explained symptoms, from digestive problems, fatigue, and urinary incontinence to frequent joint dislocation and dizziness after standing up. Because no connective tissue defect has been identified*, researchers have expressed concerns that poorly explained symptoms are incorrectly attributed to hEDS. In 2017 for example, representatives of The Royal College of Paediatrics and The British Society of Paediatric and Adolescent Rheumatology wrote:
“We are highly concerned, in the absence of a definable tissue mechanism for such symptoms, that ascribing systemic symptoms to JHS/hEDS is very unlikely to be helpful to the patient and family, and indeed may cause harm. […] There is undoubtedly strong disagreement regarding the diagnosis of hEDS (indeed we welcome further debate) but what is irrefutable is that there is currently no evidence that this is a collagen disorder (in children or adults) or that it is related to multisystem disease.”
JHS refers to joint hypermobility syndrome, a diagnosis very similar to hEDS. A group of researchers led by Rodney Grahame and Alan Hakim in the UK have made the case that symptomatic joint hypermobility should be seen as a ‘forme fruste’, an attenuated manifestation, of connective tissue disease. The prevalence of JHS is much higher than hEDS, with some reviews claiming that 3% of the population is affected by it. The 2017 diagnostic criteria discarded the JHS diagnosis because it strongly overlaps with hEDS. hEDS now takes precedence, and patients with symptomatic joint hypermobility not meeting the hEDS criteria are given the label of hypermobility spectrum disorder (HSD).
Both diagnoses assume that generalized joint hypermobility signals a connective tissue disorder that causes multiple symptoms and physical abnormalities. This theory is problematic because hypermobility is common in the general population and often asymptomatic. The evidence that it is related to pain or other symptoms is unconvincing.
Beighton score
Before we move on to discussing the epidemiological evidence, we should first say a few words about what generalized joint hypermobility is and how it is measured. The Beighton score (0-9) is by far the most used tool. In brief, it assesses:
- If you can bend your little finger backward beyond 90 degrees,
- If your thumb touches your forearm when bending your wrist,
- If your elbows bend backward beyond normal straightness (-10 degrees or more),
- If your knees bend backward beyond normal straightness (-10 degrees or more),
- If you can reach the floor with the palms of your hands while keeping your legs straight.
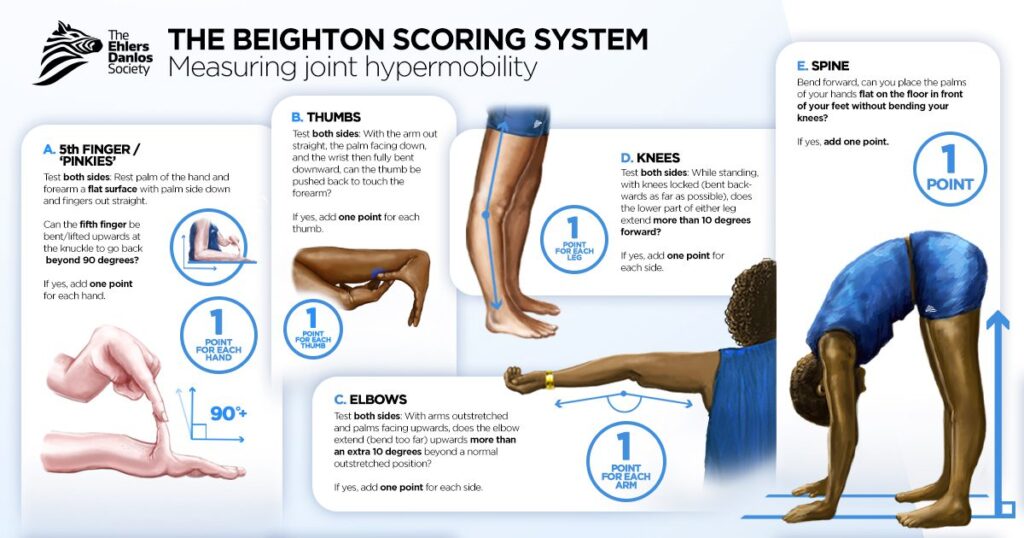
For the first four, you get a separate point for each body side the statement applies to, resulting in a maximum Beighton score of 9 points. Videos of how researchers apply the Beighton scoring system can be viewed here and here.
Depending on your age and the diagnostic criteria, Beighton scores of 4, 5, or 6 have been used to indicate generalized joint hypermobility. Notice that people who meet this threshold may have hypermobility in different joints: it does not capture a homogeneous population, as there are multiple ways to reach the threshold. Another issue with the Beighton score is that it is ‘top heavy’: it mostly assesses the upper limbs while disregarding the lower body and major joints such as the hips. And perhaps most importantly, it does not assess the severity or intensity of joint hypermobility: it simply gives a point if one of the assessments is positive.
The Beighton score was developed 50 years ago as a rapid screening tool for hypermobility in South Africa. Today, the main justification for its use seems to be that it has already been used a lot. It remains, however, the most commonly used tool in research and the method that all diagnostic criteria rely on. Therefore, we will have to temporarily overlook its shortcomings to move to the main subject of this blog: is there a link between hypermobility and hEDS/JHS symptoms such as musculoskeletal pain?
No link between hypermobility and pain
To answer that question, we need epidemiological studies that examine a representative sample of the general population. Most of these studies have been conducted in children or young adolescents and found that generalized joint hypermobility is more common in young, female, and non-Western populations. Studies in African and Asian countries found higher prevalence rates of joint hypermobility than European studies and some point to a correlation with factors such as malnutrition. Results are therefore often grouped by ethnicity to reduce heterogeneity.
A 2012 systematic review found no association between hypermobility and musculoskeletal pain in American and European studies (Odds ratio: 1.00 [0.79 -1.26]). We also made an overview of epidemiological studies on children and adolescents in Western countries (Table 1) and came to the same conclusion: most studies show no relationship. If you divide a population in two based on whether they have generalized joint hypermobility, the frequency of arthralgia (joint pain) and musculoskeletal pain is roughly the same in both groups. One Italian study, for example, reported that “no association was found between hypermobility and musculoskeletal pain. Hypermobile children did not experience functional limitations in daily activities, and they were slightly more active than non-hypermobile children.”
| Study | Outcome | Population | Findings |
| Fairbank 1984 | Back pain | N = 446 13-17 year olds UK | 26% had back pain. Femoral and tibial rotation were significantly less in the pupils with back pain. No difference was detected in the upper limb measurements of joint mobility of the pupils with and without back pain. |
| Gedalia 1985 | Juvenile episodic arthritis/arthralgia (JEA) | N =260 5-17 year olds USA | Different cohorts with different selection criteria were used. Hypermobility was more comon in JEA (66%) than in controls (12%) and rheumatoid arthritis (3%). |
| Arroyo 1988 | Arthralgia | N = 192 5-19 year olds USA | 34% were hypermobile and 50% of the hypermobile group had a history of arthralgia, compared to 20% of controls. 10% in each group had arthritis. |
| Gedalia 1991 | Recurrent arthralgia | N = 429 6-11 year olds Israel | 12% had hypermobility. Recurrent arthralgia occurred at a higher frequency (40%) in the hypermobile group than in the non-hypermobile group (17%). |
| Mikkelsson 1996 | Pain at least once per week | N = 1637 Pre-adolescents Finland | 7.8% had a Beighton score ≥ 6. 29.9% with hypermobility had pain compared to 32.3% in controls. Hypermobility in one joint was not associated with pain in that body area. Disability did not correlate with the total Beighton score. |
| Harreby 1999 | Low back pain (LBP) | N = 1389 13-16 year olds Denmark | 14% hypermobility, 19.4% had LBP. “General hypermobility at any level […] was not correlated with LBP in any degree.” |
| Qvindesland 1999 | Pain | N = 267 12 year olds Iceland | 27.7% had Beighton ≥ 4. 55% of hypermobile patiens reported pain versus 52% of controls. |
| Ruperto 2004 | Functional ability (CHAQ) and well-being (CHQ) including bodily pain | N = 311 6-19 year olds Italy | 34% had hypermobility using Beihgton ≥ 5. “The presence of joint hypermobility does not affect the functional ability and the physical and psychosocial well being of otherwise healthy children.” |
| Leone 2009 | Musculoskeletal pain, disability and physical activity | N = 1230 7-15 year olds Italy | 22.2% had Beighton ≥ 5. 22% of children with musculoskeletal pain versus 23% of controls had hypermobility (OR 1.057, 95% CI 0.7 to 1.4). “Hypermobile children did not experience functional limitations in daily activities, and they were slightly more active than non-hypermobile children” |
| Juul-Kristensen 2009 | Musculoskeletal complaints | N = 411 8 year olds Denmark | “29% of the children had GJH4, 19% had GJH5, 10% had GJH6, and 9% had BJHS. There was no difference in daily level and duration of physical activity and in frequency of musculoskeletal pain and injuries between those with and without GJH.” |
| Smits-Engelmans 2011 | Pain in joints, muscles, or ligaments | N = 551 6-12 year olds Netherlands | 35% had Beighton ≥5, 12.5% had joint pain which was lower in those with high hypermobility 64% Beighton<5: 13.3% joint pain26.5% Beighton<7>5: 12.8% joint pain9.1% Beighton>7: 4.1% joint pain |
| Sperotto 2014 | Chronic musculoskeletal pain (MSP) | N = 289 8-13 year olds Italy | 13.2% had Beighton≥4. 32% of hypermobile had MSP compared to 29% in controls (p=0.054). |
| Aartun 2016 | Neck and back pain | N = 963 11-15 year olds Denmark | 6.5% had Beighton score ≥ 5 and 4.25% ≥ 6. Neck and back pain occurred > 50%. Hypermobility had no significant association or predictor value for pain. |
| Morris 2016 | Number of pain sites | N = 1584 14 year olds Australia | 48% had Beighton≥4 and 18.6% Beighton≥6. In boys there was an association with hypermobility and more than 3 pain sites, but not in girls and only for Beighton≥6 threshold. No associations were found for number of pain areas lasting >3 months for either boys or girls at either threshold. |
Several studies followed patients over time and reported hypermobility to be a predictor of future pain, but they come with important caveats. A Danish study, for example, found that pain was significantly worse in the hypermobility group, but only when defined by a Beighton score of 4 or higher, and not when threshold of 5 or 6 was used. There were also no significant differences for quality of life and overall symptoms between the hypermobile and control group. A Finnish study found a relationship between pain and hypermobility but only at the 4-year follow-up, not at baseline or 1-year follow-up. A UK study reported that joint hypermobility increases the risk of moderately troublesome musculoskeletal pain at the shoulder, knee, and ankle but not in other sites such as the spine, elbows, hands, and hips. The effect sizes were relatively small (odds ratios between 1.68 and 1.84), and when the authors looked at pain intensity scores or pain interfering with daily activities, they found no significant differences. A more recent Danish study reported that joint hypermobility had no predictive value for neck and back pain.
Studies on adults are scarce. Two of the largest studies used a 5-item questionnaire to assess joint hypermobility. Because participants had to assess hypermobility themselves, these studies are at risk of various reporting biases (e.g., participants who report symptoms might be more likely to also report other conditions such as hypermobility). One survey in the UK found that hypermobile individuals were significantly more likely to report pain, but the difference was small: 18.5% versus 15.8% in controls. In the statistical models used, hypermobility accounted for less than 1% of the variability in pain. The authors concluded: “The relationship was relatively modest and may be explained by unmeasured confounding factors…” A similar questionnaire-based study was done in Denmark with 1006 participants. 30% of participants had hypermobility, as assessed with the 5-item questionnaire. The quality of life score was lower in hypermobile people than in the control group, but the difference was small: a score of 80 versus 85 out of 100. A third study assessed the 0-9 Beighton score in young adults in Florida. The authors report that “there was no difference in the proportion of respondents who reported chronic joint pain across the three Beighton score categories (11.9%, 16.1%, and 12.9% of respondents with Beighton scores of 0, 1–4, and 5–9, respectively)”.
In conclusion, there is currently no reliable evidence that hypermobility is associated with pain and disability. Most studies found no or only a small to negligible difference in pain outcomes between those who are hypermobile versus those who are not. The literature, however, is limited, especially in adults, where the largest studies rely on self-assessment of hypermobility. Most studies also focus on the proportion reporting pain rather than pain intensity or more specific measures of disability.
Furthermore, the studies listed above show that hypermobility and widespread pain are common. In an Australian study, 18.6% of adolescents had generalized joint hypermobility, even when a Beighton score of 6 or higher was used as the threshold. Chronic widespread pain also has a prevalence of approximately 10% in the general population. Women are more likely than men to have hypermobile joints and widespread pain, but even if we assume no relationship between the two, there will be a large group of patients with both hypermobility and musculoskeletal symptoms. It is not uncommon for physicians to see many such patients in their clinic. This does not mean that hypermobility and pain are related. Diagnoses such as JHS and HSD that are based on that assumption lack a solid evidence base.
hEDS systemic features
The diagnostic criteria for hEDS are more complex as they require three requirements to be met (see below). The first criterion is generalized joint hypermobility and the third involves excluding other diseases that might explain the symptoms. The second criterion asks about various signs and symptoms and consists of three parts: A, B, and C, two of which must be present.
Having widespread pain, which is the case for approximately 10% of the population, fulfills part C. Part B is met if a first-degree relative (parent, sibling, or child) has hEDS which may be the case for some because both joint hypermobility and pain have a genetic component. However, most individuals being evaluated for an hEDS diagnosis will need to meet part A. It requires the presence of 5 out of 12 clinical signs and abnormalities.
Table 2: 2017 hEDS criteria
The clinical diagnosis of hypermobile EDS needs the simultaneous presence of all three criteria.
Criterion 1—Generalised joint hypermobility
Beighton hypermobility score: ≥6 in pre-pubertal children and adolescents ≥5 in pubertal men and woman to age 50 years ≥4 in men and women over the age of 50 years.
If Beighton score is one point below age- and sex-specific cut-off, two or more of the following must also be selected to meet criterion:
- Can you now (or could you ever) place your hands flat on the floor without bending your knees?
- Can you now (or could you ever) bend your thumb to touch your forearm?
- As a child, did you amuse your friends by contorting your body into strange shapes or could you do the splits?
- As a child or teenager, did your shoulder or kneecap dislocate on more than one occasion?
- Do you consider yourself “double jointed”?
Criterion 2—Two or more of the following features (A, B, or C) must be present:
Feature A (five must be present):
- Unusually soft or velvety skin
- Mild skin hyperextensibility
- Unexplained striae distensae or rubae at the back, groins, thighs, breasts, and/or abdomen in adolescents, men, or pre-pubertal girls without a history of significant change in body fat or weight
- Bilateral piezogenic papules of the heel
- Recurrent or multiple abdominal hernia(s)
- Atrophic scarring involving at least two sites and without the formation of truly papyraceous or haemosideric scars as seen in classical EDS
- Pelvic floor, rectal, and/or uterine prolapse in children, men, or nulliparous women without a history of morbid obesity or other known predisposing condition
- Dental crowding and high or narrow palate
- Arachnodactyly, as defined by one or more of: Positive wrist sign (Walker sign) on both sides, Positive thumb sign (Steinberg sign) on both sides
- Arm span-to-height ratio ≥1.05
- Mitral valve prolapse mild or greater based on strict echocardiographic criteria
- Aortic root dilatation with z score ≥2
Feature B:
- Positive family history; one or more first degree relatives independently meeting the current criteria for hypermobile EDS
Feature C (must have at least one):
- Musculoskeletal pain in two or more limbs, recurring daily for ≥3 months
- Chronic, widespread pain for ≥3 months
- Recurrent joint dislocations or frank joint instability in the absence of trauma
Unfortunately, these features comprise a miscellaneous collection that lacks justification. Some features, such as arachnodactyly, are suggestive of Marfan syndrome, another connective tissue disorder. Other features were once reported to be more common in hEDS than controls, but the evidence is underwhelming. Piezogenic papules, the small fat bumps on the back of the heel, form a good example. The hEDS criteria reference a single study from the 1980s that reported more piezogenic papules in hEDS patients than in controls, but this was only the case for painful piezogenic papules larger than 0.2 cm in diameter. This seems like an error or misunderstanding by the authors of the 2017 hEDS criteria. Bilateral piezogenic papules on the heel are not a sign of a connective tissue disorder. Two studies found that the vast majority (76%-80%) of the general population has them.
Other features are more prevalent than their medical vocabularies suggest. Dental crowding, for example, occurs in approximately 30% to 60% of children and adolescents. Unexplained striae or stretch marks are often seen in healthy adolescents and young adults, with one study finding it in 11% of young men. Hernias affect approximately 1 in 10 people during their lifetime. A Russian study, for example, found abdominal hernias in 20% of the population with recurrence rates ranging from 30% to 80% after hernia repair. Atrophic scarring is seen in up to 87% of patients with mild to moderate acne. A review found that almost half of acne patients had scars, the majority (78%) of which were atrophic. Mitral valve prolapse is also a common disorder, afflicting 0.6% to 2.4% of the general population and it is usually not clinically significant in patients with hEDS. The evidence on whether mitral valve prolapse is more common in hEDS patients is conflicting, as the 2017 criteria acknowledge.
Other features such as ‘unusually soft or velvety skin’, ‘narrow palate’, or ‘mild skin hyperextensibility’ are largely subjective. A Chilean study, for example, found 95% of control subjects (paramedical personnel) to have skin abnormalities. The authors report: “Skin abnormalities were seen in almost all patients and all controls; some of these were nonspecific, but others were typical, such as soft, lax, velvety skin and poor cicatrization (cheloids and papyraceous scars).” Mild skin hyperextensibility is also nonspecific, with one study finding it in 20% of the control group.
Some studies have reported no relationship between the features mentioned above and joint hypermobility, stating that “Higher BS [Beighton Scores] did not correlate with increased number of systemic manifestations.” If both were manifestations of the same connective tissue disease, we would expect to see a clear relationship.
Inflated concepts
In summary, the hEDS diagnosis is based on nonspecific features that are common and often benign in the general population. Patients with hypermobility, chronic widespread pain, and five of the features listed above can be diagnosed with hEDS and told that their symptoms are due to a rare connective tissue disease. This is likely not the case for most of them.
Based on the diagnostic requirements we can expect the prevalence of hEDS to be much higher than the commonly cited estimate of 1 in 5,000. Unfortunately, no study ever tested this. There is not a single epidemiological study that assessed how many people meet the hEDS criteria.** We only have smaller studies that provided clues of an inflated prevalence. One study, for example, found hEDS to be present in 18% of patients with myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) but also in 8% of the ‘healthy’ control group (the difference was not statistically significant). In a letter to the BMJ, a physician noted:
“Over the past 15 years, my caseload of patients with Ehlers-Danlos syndromes (EDS) has increased from two a year to two a month […] Overall, 40% of women could present with hypermobile EDS if they choose to do so. This does not make it a useful diagnosis.”
It is also remarkable how the range of symptoms attributed to JHS/hEDS has increased over the years. The hypermobility syndrome was originally defined as a benign disorder with mild and self-limiting musculoskeletal symptoms. Similarly, hEDS was considered a “merely musculoskeletal disorder with mild cutaneous complications”. Orthostatic intolerance or debilitating fatigue were not mentioned until publications by Rowe et al. 1999 and Voermans et al. 2010 highlighted them as important symptoms. Today JHS and hEDS encompass a wide range of complaints and disorders including gastrointestinal problems, postural orthostatic tachycardia, mast-cell activation, craniocervical instability, small fiber neuropathy, muscle stiffness, tension headaches, behavioral disturbances, and pelvic floor problems.
It could be that recognition of those symptoms has increased over the years. Some even proposed that hEDS is the “unifying concept for various functional somatic syndromes” or “the occult underlying diagnosis for many chronic somatic pain syndromes.” A more plausible explanation is that researchers have inflated the hEDS diagnosis by including patients with unexplained symptoms and no clear indication of connective tissue disease.
A Belgian study found that orthostatic and gastrointestinal complaints were significantly more prevalent in hEDS compared to classical and vascular EDS types. The authors concluded that “Remarkably, the autonomic symptom profile of EDS‐HT [an older name for hEDS] was more similar to fibromyalgia than to the other EDS groups.” Indeed, based on its symptom profile, prevalence, female predominance, and lack of molecular basis, hEDS resembles fibromyalgia and ME/CFS more than other EDS types.
| Other EDS types | hEDS | |
| Prevalence | (Very) rare: < 0.005%. | Quite common > 0.02% |
| Cause | Known genetic mutation involving extracellular matrix proteins. | Molecular basis unknown, no evidence of connective tissue disease. |
| Clinical features | Specific clinical signs such as blue sclerae, arterial rupture, extreme hyperextensible skin, pseudo tumors, etc. | Common or vague features such as joint hypermobility, widespread chronic pain, soft skin, stretch marks, dental crowding etc. |
| Female predominance | Ca. 50-60% are female | Ca. 90% are female |
| Symptoms | Often related to physical abnormalities such as kyphoscoliosis, organ rupture, easy bruising, etc. | Wide range of highly debilitating but unexplained symptoms, similar to ME/CFS and fibromyalgia. |
No correlation
Rheumatologists are often skeptical of the clinical value of hypermobility. In a 2001 survey of British consultant rheumatologists, nearly half of the respondents were doubtful about the impact of hypermobility syndromes on people’s lives, and three-quarters were skeptical of a significant contribution to the overall burden of rheumatic disease.
Studies in other conditions suggest hypermobility often has limited clinical value. For example, a study on functional gastrointestinal disorders found that “there was no significant difference in prevalence of orthostatic intolerance, OH [orthostatic hypotension], and POTS [postural orthostatic tachycardia syndrome] between those with joint hypermobility and those without.” Studies on fibromyalgia found no difference in pain intensity between patients with or without hypermobility and no correlation between Beighton scores and tender points. Another study on functional pain disorders found autonomic function to be similar in patients with and without hypermobility. A study on stomach problems reported: “We did not identify any specific differences in gastric sensorimotor function between patients with and without JHS.”
ME/CFS studies show similar results. Although some studies found increased orthostatic problems in ME/CFS patients with hypermobility versus those without hypermobility, most (examples here, here, here, and here) did not find significant differences. One study for example concluded that “the clinical importance of generalized joint hypermobility is questionable” and that “no evidence supporting the clinical importance of hypermobility in patients with CFS was provided.” Another study concluded that joint hypermobility “was not associated with other clinical characteristics of the illness.” Admittedly, we cannot conclude much from such studies, but they do suggest that hypermobility and hEDS are unlikely to be the unifying concept for functional syndromes.
Similarity to POTS
The problem with hEDS is similar to that of POTS (see here for our previous blog post on POTS). Both diagnoses are based on an underlying assumption of what causes the symptoms of patients, namely a connective tissue disorder or autonomic dysfunction. This contrasts with syndromes such as ME/CFS, irritable bowel syndrome, or fibromyalgia, which describe a clinical picture without making assumptions about their underlying causes.
While POTS was used as a left-over category for orthostatic symptoms without clear autonomic dysfunction, hEDS is the sole EDS type that has no identified genetic mutation. The literature on POTS and hEDS also lacks epidemiological studies that involve representative samples of the general population. Hallmark features of hypermobility and orthostatic tachycardia are more common and benign than either diagnosis suggests (both signs are also more prevalent in young females which may explain why some believe there is a connection between POTS and hEDS).
We hope that the connective tissue hypothesis pans out for at least a subgroup of patients with hEDS and that the ongoing HEDGE (Hypermobile Ehlers-Danlos Genetic Evaluation) study will identify new genetic mutations. However, as long as there is no robust evidence, diagnosing hEDS may be akin to putting the cart before the horse.
POTS and hEDS are both based on speculative theories without robust evidence that are rarely questioned because they are masked as an already established diagnosis. Questioning the underlying theory behind POTS or hEDS, therefore, feels like dismissing the diagnosis itself which some patients find distressing or insulting. We believe, however, that it is important to critically assess the evidence because if these theories are wrong, patients are the ones who pay the price for these mistakes.
hEDS and POTS are certainly not the only diagnoses that operate in this manner. Other examples are Mast Cell Activation Disorder (MCAS), Central Sensitivity Syndrome, Chronic Lyme Disease, and Bodily Distress Syndrome. In future blog posts, we hope to look more closely at this phenomenon of speculative theories wrapped up as diagnoses.
Notes
* An interesting 2024 study found evidence of a common extracellular matrix fragmentation pattern in the plasma of hEDS and HSD patients that was not seen in other EDS types or rheumatic diseases. The findings, however, have not been replicated. For a critical discussion of this paper see the Science for ME forum.
** hEDS prevalence estimates are based on electronic registries that routinely collect primary care and hospital admissions data. These may underestimate hEDS and JHS prevalence if patients remain undiagnosed.
This is unbelievably stupid and unscientific. You really ought to have assessed this for limitations before publishing. This is embarrassing. You are inferring there is no connective tissue disorder or disability/pain in hEDS because by your own admission, there is insufficient research? You’ve never spoken to someone with EDS obviously. Honestly this entire website is a joke.
I think you misunderstood because we didn’t say that there is no disability/pain in hEDS, quite the contrary. We highlight that there is insufficient evidence that this disability/pain is due to a connective tissue disorder. In other words, the disability/pain might have another cause and hEDS might be over-diagnosed.
>“The relationship was relatively modest and may be explained by unmeasured confounding factors such as psychological distress.”
Where is your evidence for this? I thought you were a skeptic.
It’s a direct quote (hence why it is in Italics) from the article being discussed namely:
Mulvey MR, Macfarlane GJ, Beasley M, Symmons DP, Lovell K, Keeley P, Woby S, McBeth J. Modest association of joint hypermobility with disabling and limiting musculoskeletal pain: results from a large‐scale general population–based survey. Arthritis care & research. 2013 Aug;65(8):1325-33.
https://pubmed.ncbi.nlm.nih.gov/23401475/
It’s unclear what the authors mean by psychological distress but we think that reporting biases may limit the value of studies that used self-assessed hypermobility. As we wrote: ‘Because participants had to assess hypermobility themselves, these studies are at risk of various reporting biases (e.g., participants who report symptoms might be more likely to also report other conditions such as hypermobility).’
You could have been more critical of the line you quoted. Perhaps stating that there is no convincing body of objective evidence proving psychosocial factors cause or perpetuate unexplained physical syndromes such as Hypermobility EDS or ME.
My advice to you is to be less ambiguous.
Based on your comments, we have removed the piece of the quote that mentions psychological distress. It now reads: The authors concluded: “The relationship was relatively modest and may be explained by unmeasured confounding factors…”
This is very good. You do something unique, insightful and very interesting. I’m impressed.
Questioning the dogma is always good, sometimes even after theories become laws. But seeing your daughter bend her thumb to touch her forearm for the first time during a medical appointment is pretty powerful. She has pretty much all the symptoms mentioned. As a scientist, having also reviewed the available studies, I think we simply haven’t found the gene/genes or other biochemical cause of hEDS yet. Before Pasteur we thought bad odors killed people. Now we know better.
Thanks for your comment. Hopefully the HEDGE study will provide new gene mutations and explanations for a subgroup of hEDS patients. But we also think it is likely that hEDS led to overdiagnosis of patients who may not have a connective tissue disorder as the cause of their symptoms.